Research
Biological sequences and structures
Statistics and Machine Learning for Population Genetics
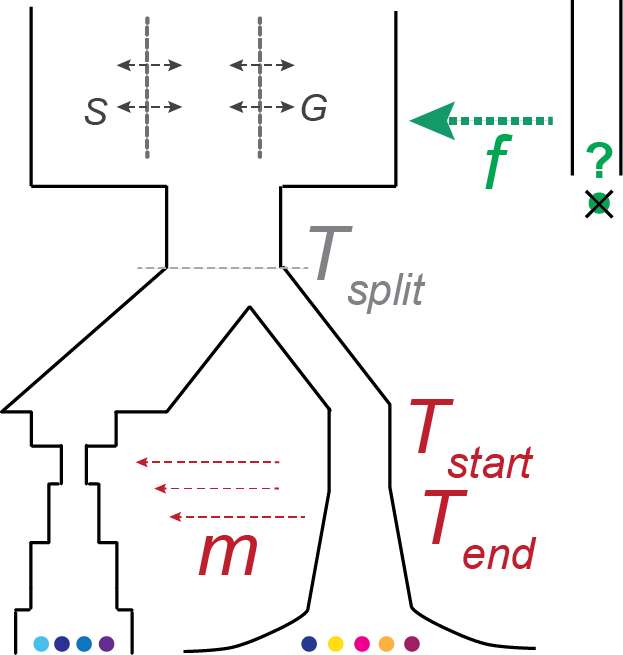
Our main expertise is the inference of population past histories using various statistical tools, from basic descriptive statistics to Bayesian Computation. It includes reconstructing the demography and relationships of human or non human species both from modern and ancient DNA sequences.
We pursue our research on the development of demographic inference methods, focusing on deep learning approaches and paleogenetics applications. Our goal is to develop a family of methods that automatically solves population genetics tasks. For that we leverage the strength of ABC to tackle highly complex tasks (corresponding to model with unknown likelihood), and the strength of deep learning to automatically extract relevant information from data. We also direct our methodological development efforts towards integrating temporal and lower quality data into population genetic methods because these are key characteristics of paleogenomics studies.
Main investigator: Flora Jay
Algorithmics of Molecular Structures
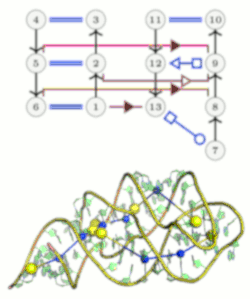
Regarding structural bioinformatics, we mainly focus on RNA structures, aiming to develop combinatorial and graph-based approaches for solving key problems in structural bioinformatics. We notably develop new approaches for the comparison and the design of RNA structures, as well as for predicting RNA-protein interactions. Meanwhile we mainly focused on analysis and prediction of RNA three-dimensional (3D) structures.
Our main long-term objective is to better understand how RNA folds, and to develop data-driven algorithmic approaches able to predict the structure of large RNA molecules. Our works on RNA structure alignment, on coarse-grained sampling on 3D structures, and on fine-tuned analysis of recurrent interaction motifs, give us a solid basis to go further towards the prediction of RNA 3D structures, given the sequence only.
Main investigators: Alain Denise, Christine Froidevaux, Loïc Paulevé
Computational Systems Biology
Modelling and simulation of biological networks
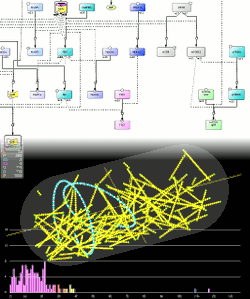
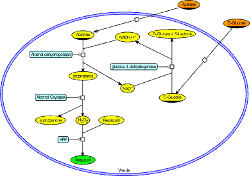
We are developing logic-based methods that allow to build and complete signaling networks. Construction of the network model is performed by means of deductive reasoning using formalised expert rules applied on experimental results obtained from various types of experiments, while completion is performed by abductive reasoning. Our work is based on Systems Biology standards such as the Systems Biology Graphical Notation (SBGN), that give a starting point and a framework of thought to our methods.
Many biological processes involve a small number molecules, where their spatial localisation is the key cause of the behaviour of the whole system. The HSIM simulation system implements hybrid simulations mixing entity-centered model that can take into account spatial localisation, explicit diffusion, molecular crowding and molecular aggregates with an efficient approximated Stochastic Simulation Algorithm (SSA) for numerous and homogeneously localised molecular species. An integrated ODE solver (adaptative RK4 method) is also provided within the Hsim system.
Keywords: model abduction, logic programming; entity-centered models, stochastic processes
Main investigators: Christine Froidevaux, Patrick Amar
Main software: HSim
Steady-state and dynamics analysis for biological networks
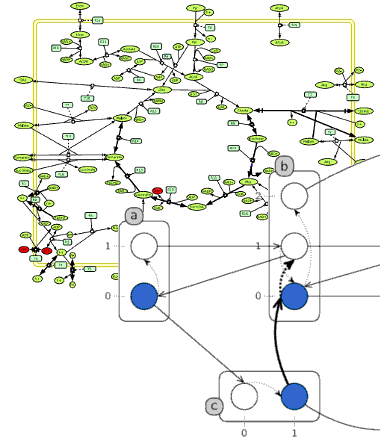
Elementary flux modes (EFMs) are the smallest sub-networks enabling the metabolic system to operate in steady state. In order to improve the scalabiliy and accuracy of such an analysis, we are developping new methods for computing EFMs based on SMT solvers and by integrating relevant biological constraints.
For signalling pathways and regulatory networks, we are developing new methods for capturing the transient dynamics of large-scale Boolean and discrete networks (up to several thousands nodes). With the design of abstractions of causality in automata networks, we provide efficient algorithms for analysing reachability properties and deducing potential mutations (cut sets) for controlling their emergence in biological networks.
Keywords: elementary flux modes, metabolic networks; automata networks, abstract interpretation
Main investigators: Sabine Peres, Loïc Paulevé
Main software: Pint
Synthetic biology
Synthetic biology studies how to design and construct biological systems with functions that do not exist in nature. Biochemical networks, although easier to control, have been used less frequently than genetic networks as a base to build a synthetic system.
We describe a methodology for the construction of synthetic biochemical networks based on three main steps: design, simulation and experimental validation. We developed NetDraw to help users to go through these steps. NetDraw is a SBGN compliant editor that allows the design of abstract networks that can be implemented thanks to MetaGate, an automated logic gate extractor from metabolic networks of living organisms. NetDraw enables also simulations with an integrated version of the HSIM simulator.
A direct application of these principles is the design ab initio of a synthetic biochemical network embedded in a lipid vesicle, in order to detect a pattern of biomarkers specific of a target pathology (e.g. diabetic nephropathy)
Keywords: synthetic biology; enzymatic networks; SBGN
Main investigator: Patrick Amar
Main softwares: NetDraw, MetaGate
Data integration
Scientific workflows
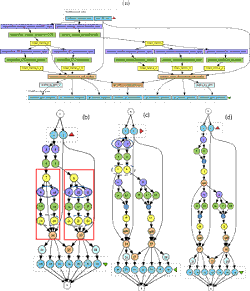
Bioinformatics experiments are increasingly complex, involving very large sets of interrelated tools and making use of huge data sets. Scientific workflow management systems have been designed to help users design and execute their experiments. Supporting scientists in designing reproducible and reusable workflows and managing the execution of such large-scale experiments is of paramount importance to enhance new discoveries in Biology. Our areas of interest include optimizing workflows (e.g., removing unnecessary steps), querying workflow repositories (e.g., retrieving similar workflows), and managing provenance information (e.g., understanding differences between two executions). Our approaches make use of database and graph-based techniques.
Keywords: workflow similarities, provenance, database
Main investigators: Sarah Cohen-Boulakia, Christine Froidevaux
Main software: DistillFlow
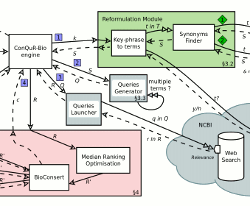
Biological data ranking
Another challenge is to help scientists face the deluge of data available. The problem is actually two folds: the number of biological databases available is increasing over time and the number of answers returned by even one database may be too large to be dealt with. A first series of approaches we propose aim at guiding users in the maze of biological databases while considering user preferences in the kind of resources to be queried. As for ranking biological data, the main difficulty lies in taking into account various ranking criteria and combine them. Our originality lies in considering rank aggregation (a.k.a. median ranking). Our approaches make use of database and combinatory techniques.
Keywords: database, consensus ranking, combinatorics
Main investigators: Sarah Cohen-Boulakia, Alain Denise, Christine Froidevaux, Adeline Pierrot
Main softwares: ConQuR-Bio, GeneValorization